Field Note 11: Dispatch - HTGAA Week 4 Homework

Part B: Protein Analysis and Visualization
Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
Briefly describe the protein you selected and why you selected it.
Tyrosinase is an enzyme that plays a crucial role in the production of melanin pigment. As part of my interest in producing different colors and pigments using synthetic biology, I selected tyrosinase and the melanin pathway as my focus for this phase becuase it is a one enzyme pathway and should be relatively simple to work with.
Identify the amino acid sequence of your protein.
MSNKYRVRKNVLHLTDTEKRDFVRTVLILKEKGIYDRYIAWHGAAGKFHTPPGSDRNAAHMSSAFLPWHREYLLRFERDLQSINPEVTLPYWEWETDAQMQDPSQSQIWSADFMGGNGNPIKDFIVDTGPFAAGRWTTIDEQGNPSGGLKRNFGATKEAPTLPTRDDVLNALKITQYDTPPWDMTSQNSFRNQLEGFINGPQLHNRVHRWVGGQMGVVPTAPNDPVFFLHHANVDRIWAVWQIIHRNQNYQPMKNGPFGQNFRDPMYPWNTTPEDVMNHRKLGYVYDIELRKSKRSS
How long is it? What is the most frequent amino acid?
The Tyrosinase protein is 297 amino acids long. The most frequent amino acid is Proline (P), which appears 22 times.
How many protein sequence homologs are there for your protein?
Against the ClusteredNR database, there are 100 clusters of homologous sequences, some clusters range from 1-12 members, with a cluster contaning 166 members being the largest.
Does your protein belong to any protein family?
Tyrosinase belongs to the Tyrosinase and Hemocyanin family and the Di-copper centre-containing domain superfamily
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure?
The structure of Tyrosinase was deposited on 22nd June 2010. It is of good quality with a resolution of 2.00 Å.
Are there any other molecules in the solved structure apart from protein?
Yes, It contains Copper (II) ions, Zinc ions, Chloride ions & Water molecules (HOH).
Does your protein belong to any structure classification family?
According to SCOP/SCOPe Classification it is classified as All alpha proteins and according to CATH Classification it belongs to the Mainly alpha class.
Open the structure of your protein in any 3D molecule visualization software
Color the protein by secondary structure. Does it have more helices or sheets?
It has more helices than sheets, I have colored the Alpha Helices in red and the Beta Sheets in yellow.

Structural View of Tyrosinase (Red Alpha Helices, Yellow Beta Sheets)
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Hydrophilic Residues (in Blue) are more abundant than the Hydrophobic Residues (in Red).
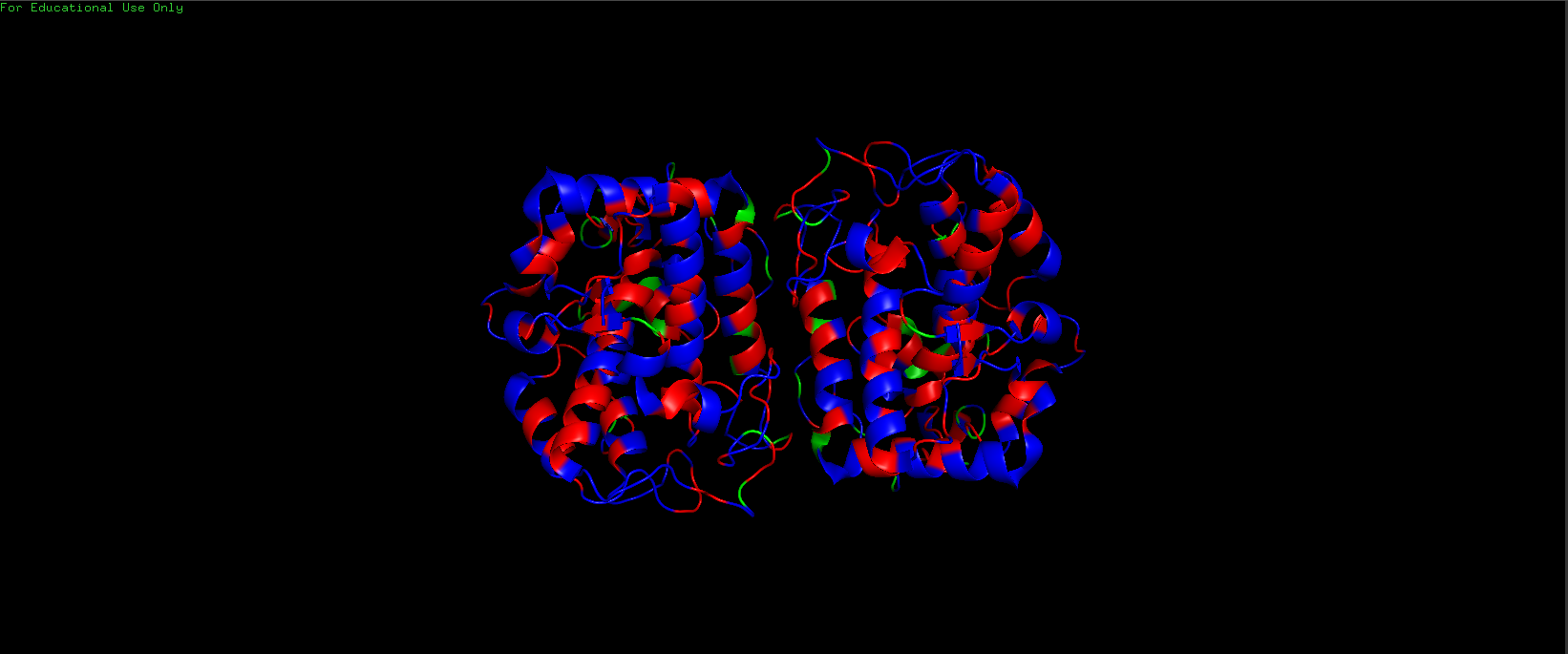
Hydrophilic (Blue) vs Hydrophobic (Red) Residues in Tyrosinase
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
It has 2 binding pockets, I have colored the Copper (II) ions in orange to highlight the location of binding pockets.
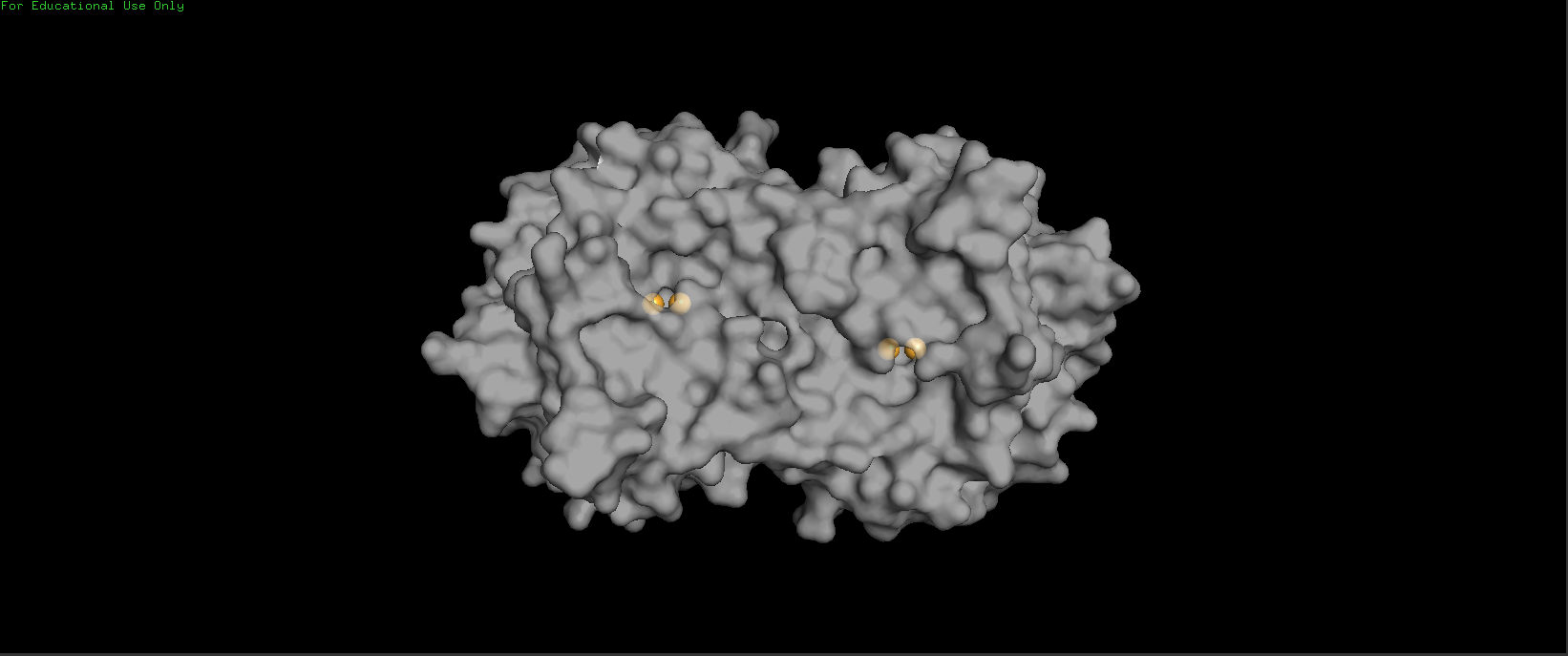
Active Sites of Tyrosinase (Copper (II) ions in Orange) - Transparency = 0.35
Part C. Using ML-Based Protein Protein Tools
Fold your protein with AlphaFold or ESMFold or Boltz and compare it to the real structure.
I folded my 297 amino acid Tyrosinase sequence using the AlphaFold web server. The resulting prediction was highly accurate and very similar to the experimental PDB structure (3NM8).
Note that i used AlphaFold Monomer for this prediction, while the Tyrosinase structure is a Dimer.

AlphaFold Prediction of Tyrosinase Monomer
Any predicted vs. experimental differences.
Apart from the Dimer vs Monomer difference, the alphafold prediction doesn’t also show the Copper (II) ions that are present in the experimental structure.
Low-confidence regions and why do you think they are low confidence?
The prediction was extremely confident, with a pTM score of 0.95. The vast majority of the structure was “dark blue,” indicating very high confidence. There were only a few small loops with lower confidence (yellow to orange), likely due to their inherent flexibility or lack of defined structure.
Inverse-fold your structure with ProteinMPNN
What sequence do you get?
>T=0.1, sample=0, score=0.7689, seq_recovery=0.4561
MSRVRKPVEDLTDEEWKKYVAAVQKLRDDGTYRKFVDDWRDAYLHKIPPGSTRNAIANGPSFIPAHIAMLYRFEKALQAVDPTVTLPYIDFDKIAELEDPSKSIVFRDDRLGGNGDAATGNKVTTGPFAAGKWETYDANGNPSGGLTRNLGASAEAPKLPTQAEVDAALKIKNYSTPPYDETSKNSFVNAIEGYENYPKLFRRILKWFGGEMATPLTAMNDPAFWLIHAYVVKVYDKWKKKHKNVSYYPKSGGPKGANYNDPIYPTDITAKDVEDLEKLGFRFAG
Is it the same as the original sequence you folded? Why yes or no?
No, it was completely different from the original (the “sequence recovery” was only 45%).
This is because the amino acid code is redundant, just like the DNA codon code. many amino acids can code for the same structural job. so 2 diffrerent amino acid sequences can fold into very similar 3D structures that have the same function or job.
Part D. Group Brainstorm on Bacteriophage Engineering
You can View the Proposal Through Here
Check out Other HTGAA Submissions from Here